
 Bryan Pollard, president of Hyperacusis Research, introduced the well-received symposium on “Auditory Nociception and Pain Hyperacusis” at the 2016 Association for Research in Otolaryngology (ARO) Mid-winter Meeting in San Diego. He began with a key slide from his first ARO presentation, in 2013, showing a knife in the ear. That’s how so many people describe their hyperacusis: Sound is not just heard, but is felt as a knife-like stabbing pain. Pictured at top: Charlie Liberman speaks at the symposium.
Bryan Pollard, president of Hyperacusis Research, introduced the well-received symposium on “Auditory Nociception and Pain Hyperacusis” at the 2016 Association for Research in Otolaryngology (ARO) Mid-winter Meeting in San Diego. He began with a key slide from his first ARO presentation, in 2013, showing a knife in the ear. That’s how so many people describe their hyperacusis: Sound is not just heard, but is felt as a knife-like stabbing pain. Pictured at top: Charlie Liberman speaks at the symposium.
At the time — just three years ago — the concept of noise-induced pain was a mystery to researchers, doctors and audiologists. Since then, Hyperacusis Research has worked diligently to assimilate ideas and research in order to identify the biological mechanisms causing this pain.

Bryan Pollard
Bryan also highlighted Hyperacusis Research’s “Roadmap to a Cure,” created last year, and the success we have had in fundraising and increasing the number of grants given to researchers. He also emphasized the most important motivation for research: The people suffering so tremendously. His slide presentation, “Faces of Hyperacusis,” showed people from around the world who suffer from hyperacusis. In most cases, this hyperacusis was noise-induced. He ended the introduction with examples of how sufferers themselves are having an impact by writing media stories (most recently on the medical news site StatNews) and by assimilating scientific knowledge into everyday language that helps patients understand the research like the website hyperacusisfocus.org.
Hyperacusis from the patients and clinicians perspective
Richard Tyler, Professor of Otolaryngology, University of Iowa

Richard Tyler and Jaime García-Añoveros
Rich Tyler started the symposium by defining “hyperacusis” and describing various types of hyperacusis. He defined hyperacusis as an abnormal, excessive response to sound. While he briefly mentioned annoyance- and fear-based hyperacusis, his focus was on loudness hyperacusis and pain hyperacusis.
In loudness hyperacusis, sounds are perceived as louder than they would be for a normal listener. In pain hyperacusis, certain sounds, even at moderate loudness levels, evoke pain. For example, clanging dishes or a passing siren can initiate a pain cycle that may last for hours or days. Patients described the pain as “burning deep in my inner ear,” a “raw” feeling or “a deep itch I can’t scratch.” These descriptions were valuable, as other speakers referred to them later in the symposium.
Rich referenced the 2015 paper “Validation of Screening Questions for Hyperacusis in Chronic Tinnitus” that showed 2/3 of hyperacusis patients experience pain.
He also noted that hyperacusis is often, but not always, present with hearing loss and/or tinnitus. A variety of causes for hyperacusis have been identified (noise exposure, head and neck injuries, Williams syndrome, Lyme disease) but sometimes the cause is unknown. Loudness hyperacusis can be measured directly, and questionnaires are available to gauge for pain. A variety of other ear symptoms often coexist with hyperacusis. Clearly, as with tinnitus, there are many subtypes of hyperacusis.
One key point is that, at present, there are no cures. Counseling and sound therapy can be helpful for some. Sound therapies use devices that provide low-level background sound. Rich concluded by emphasizing the importance of understanding individual differences and coexisting symptoms.
The cochlear nerve: type I vs. type II neurons and the phenomenon of hyperacusis
M. Charles Liberman, Professor of Otolaryngology, Harvard Medical School
Charlie (pictured at top of page) started by reviewing the historical findings of the differences between Type I and Type II nerve fibers within the cochlea. The cochlea’s inner hair cells are connected to the brain via myelinated (Type I) fibers, and the outer hair cells are connected to the brain via unmyelinated (Type II) fibers. Since the Type II fibers are not only small in caliber but few in number (composing only 5% of all hair cell fibers), their physiological responses have proven difficult to characterize. Nevertheless, aspects of their neuroanatomy have long suggested that these Type II fibers are involved in the sensation of auditory pain. Charlie described how all mammals show strong similarities in their Type I and Type II fiber design, so similar conclusions can be drawn from many different mammal studies.
Experiments have shown that there is no acoustic response when shocking Type II ganglion cells. This finding, along with other supporting data, led Charlie to the understanding that the response of Type II fibers is not driven by normal acoustical stimuli. Type II fibers also innervate another part of the cochlea called Hensen’s cells, in the apical cochlea. Hensen’s cells collapse and recover when overstimulated by noise. Therefore, overstimulation from loud noise can lead to a Type II signal response.
Charlie next showed how Type II fibers are modulated by the medial olivocochlear (MOC) efferent nerves (see diagram below). For a more detailed description of the MOC reflex, see this link. The interconnected nature of the Type II fibers with the MOC efferents demonstrates the complexity of the auditory system. This complexity has required well-designed experiments to ensure that the functions of each component can be determined for both a normal ear and a damaged ear. While some have proposed that the Type II fibers drive the MOC reflex response, Charlie showed data that did not support that theory.
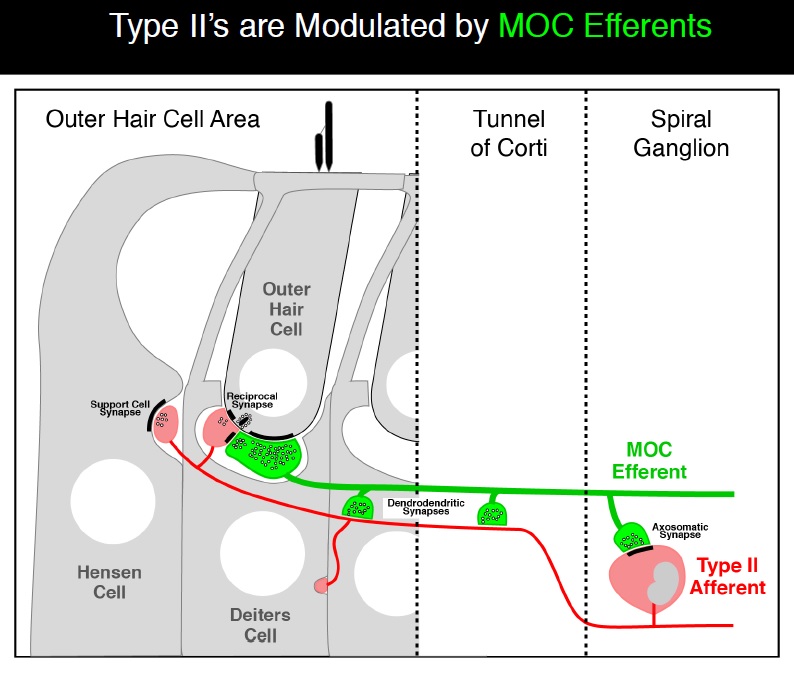
Charlie concluded by stating that Type II dysfunction may underlie hyperacusis with pain, while differences in the Type I pathway may underlie differences in loudness discomfort.
Auditory nociception, the detection of sounds harmful to the inner ear, is mediated by a non-canonical form of communication from cochlea to brain
Jaime García-Añoveros, Associate Professor, Northwestern University

Jaime García-Añoveros
Jaime introduced his talk with some key background concepts on tissue damage and nociceptors. Although cochlear hair cells are specialized for detecting sound-induced vibration, intense and persistent noise will damage and ultimately destroy them. Throughout most of the body, nociceptors of the dorsal root and trigeminal ganglia detect tissue damage or the physical stimuli causing it. However, somatosensory (relating to the perception of sensory stimuli) neurons do not innervate the organ of Corti, raising the question of whether its damage goes undetected or whether the cochlea has an alternative, nociceptor-like mechanism. The only sensory neurons innervating the organ of Corti originate from the spiral ganglion, roughly 95% of which innervate exclusively inner hair cells (IHCs). Upon sound stimulation, IHCs release glutamate to activate AMPA-type receptors on these myelinated Type I neurons, which carry the neuronal signals to the cochlear nucleus. The remaining spiral ganglion cells, Type IIs, are unmyelinated and contact outer hair cells (OHCs). Their function have previously been unknown but they resemble somatosensory neurons.
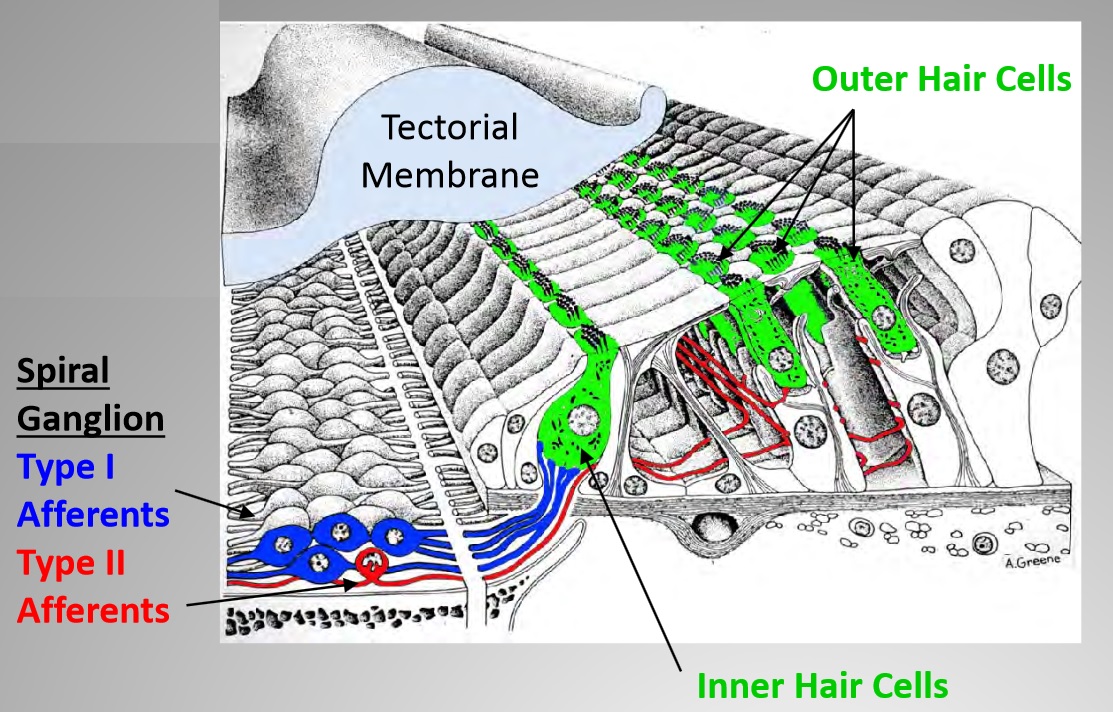
In order to demonstrate the neural connections for Type II afferents only, Jaime used mutant mice that did not have a key glutamate transporter called Vglut3, which drives IHC Type I afferents, which are the primary nerve fibers that detect noise. Jaime first documented neuronal activation in the brainstem of mice without Vglut3, in which the canonical auditory pathway (activation of Type I afferents by glutamate released from IHCs) was silenced. Next, Jaime exposed mice without Vglut3 and control littermates to ambient, innocuous (80dB) or noxious (120dB) noise and then measured neuronal activity in the cochlear nucleus and other brainstem areas.
In mice without Vglut3, Jaime found significant responses to noxious noise (120dB), which damages hair cells, but not to innocuous noise (80dB), by neurons of the cochlear nucleus, but not of the vestibular or trigeminal nuclei. Noxious noise activated neurons in the cochlear nucleus in a pattern consistent with the innervation of Type II afferents. Some areas of the cochlear nucleus activated were different than normal auditory responses. The response to noxious noise originates in the cochlea and not in other areas also stimulated by intense noise (middle ear and vestibule), as the response was absent in another type of mutant mice with selective cochlear degeneration but normal vestibular and somatosensory function.
Jaime concluded by noting that these results imply the existence of an alternative neuronal pathway from cochlea to brainstem that is activated by tissue-damaging noise and does not require glutamate release from IHCs. This detection of noise-induced tissue damage, possibly by Type II cochlear afferents, represents a novel form of sensation he termed “auditory nociception.” Jaime proposed that the auditory nociceptive system may trigger the pain sensation often associated with trauma-induced hyperacusis, since pain hyperacusis represents a pathological sensation akin to neuropathic pain. He added that auditory nociception may trigger reflexes such as middle ear muscle reflexes and neurogenic inflammation via release of peptides from vesicles in peripheral terminals to recruit immune cells.
What is the ‘Adequate Stimulus’ for Type II Cochlear Afferents?
Paul Fuchs, Johns Hopkins University School of Medicine

Paul Fuchs
Paul opened his discussion by describing the differences between Type I and Type II fibers in the mammalian cochlea. Paul’s work focuses on characterizing these fibers ex vivo (with the cochlea removed for more in-depth investigations). Type 1 afferents are the dominant, large-diameter, myelinated fibers that are postsynaptic to a single inner hair cell and carry acoustic information to the brain. The remaining thin, unmyelinated Type II afferents extend hundreds of microns along the cochlear duct to contact many outer hair cells, but are insensitive to sound.
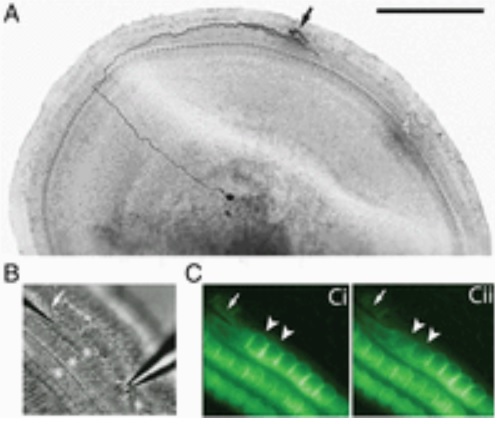 Paul described his work with intracellular recording from the peripheral process, which shows that Type II afferents are poorly activated by outer hair cell transmitter release. Outer hair cell ribbon synapses are less organized and release glutamate less efficiently in comparison to ribbons of inner hair cells. Maximal stimulation of all presynaptic OHCs is required to exceed spike threshold. So Type II afferents respond to only loud, traumatic sounds.
Paul described his work with intracellular recording from the peripheral process, which shows that Type II afferents are poorly activated by outer hair cell transmitter release. Outer hair cell ribbon synapses are less organized and release glutamate less efficiently in comparison to ribbons of inner hair cells. Maximal stimulation of all presynaptic OHCs is required to exceed spike threshold. So Type II afferents respond to only loud, traumatic sounds.
The key point of Paul’s finding is that Type II afferents are strongly activated when outer hair cells are damaged. This response depends on both ionotropic (P2X) and metabotropic (P2Y) purinergic receptors, binding ATP released from nearby supporting cells in response to hair cell damage. Selective activation of P2Y receptors (by exogenous UTP) increased Type II afferent excitability by the closure of KCNQ-type potassium channels, a potential mechanism for the painful hypersensitivity known as hyperacusis. The image above comes from the recent publication “Unmyelinated Type II afferent neurons report cochlear damage” by Chang Liu, Elisabeth Glowatzki, and Paul Albert Fuchs.
Paul concluded by stating that Type II afferents may be the cochlea’s nociceptors, prompting avoidance of further damage to the irreparable inner ear.
Auditory versus aural nociception: Evidence for partial overlap of cortical activity in humans
Ulf Baumgaertner, Heidelberg University

Ulf Baumgaertner
Ulf started by explaining the fundamental differences between the sensory system nerve pathway and the normal sensory pathway. It is evident that most painful sensations are due to activation of the nociceptive system and not overstimulation of some other sensory system (e.g. tactile or olfactory). Ulf presented the slide below to highlight the spinothalamic nerve tract which ascends from the spine to the thalamus in the brain. The spinothalamic tract is involved with perceptions of temperature, itch, touch and pain.
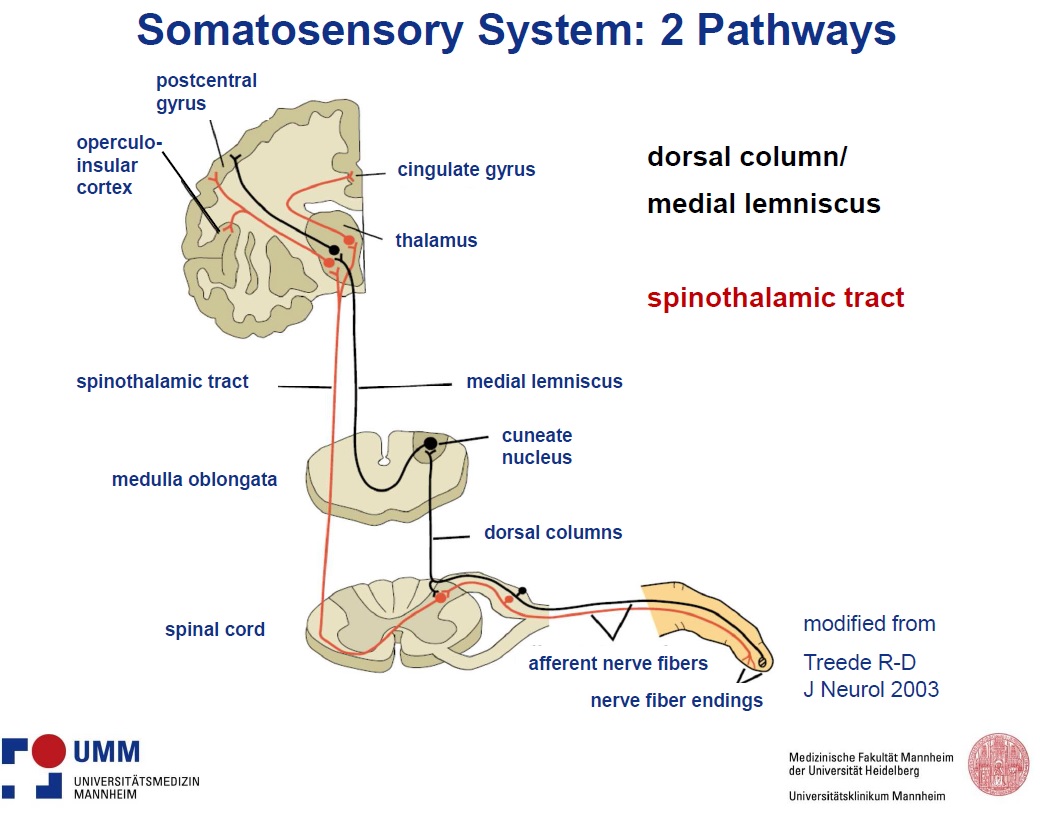
While the pain pathways for most of the body have been determined, researchers have not determined the signaling pathways that mediate pain due to intense auditory stimuli — above around 120 dB — that normal people experience. There are a few possibilities Ulf proposed as a pain pathway:
- Excitation of trigeminal nociceptive fibers of the middle ear (eardrum/tympanic cavity)
- Unpleasant pain-like feeling feeling unrelated to nociception through “standard” sensory channel (cochlear inner hair cells) converging on limbic areas (amygdala, cingulate)
- Separate high threshold sensory channel from sensory organ (cochlea)
- Combination of the above
Ulf’s team designed an innovative experiment to gain insight to the painful noise neural pathway by comparing brain fMRI images of volunteers exposed to pinpricks on the earlobes to images when the volunteers were exposed to painfully loud sounds of 130dB. Sixteen healthy volunteers (8 female, 8 male) with a mean age of 27 years participated in five separate sessions (2 auditory, 2 pinprick and 1 somatosensory). The painful auditory stimulus consisted of a 3.2 kHz tone presented at a level of 130 dB for the duration of the stimulation block (23 seconds) with irregular interruptions of 1 second in order to prevent trauma. The auditory control stimulus with the identical position of silent gaps was presented at a level of 75 dB. For somatic nociceptive stimulation, aural pinprick stimuli were applied to the ventral side of the transition zone from the lobule to the inferior helix using a custom-made clip containing 10 sharp plastic pins. Pain ratings to auditory (64/100 ± 3) and pinprick stimuli (70/100 ± 4) were similar.
Conjunction analysis of painful and non-painful auditory stimuli showed activation of the primary and secondary auditory cortex. Contrast analysis of painful vs. non-painful auditory stimuli showed activation of the medial thalamus, anterior (mid-) insular cortex bilaterally, inferior parietal lobule and supramarginal gyrus, as shown in the slide below.
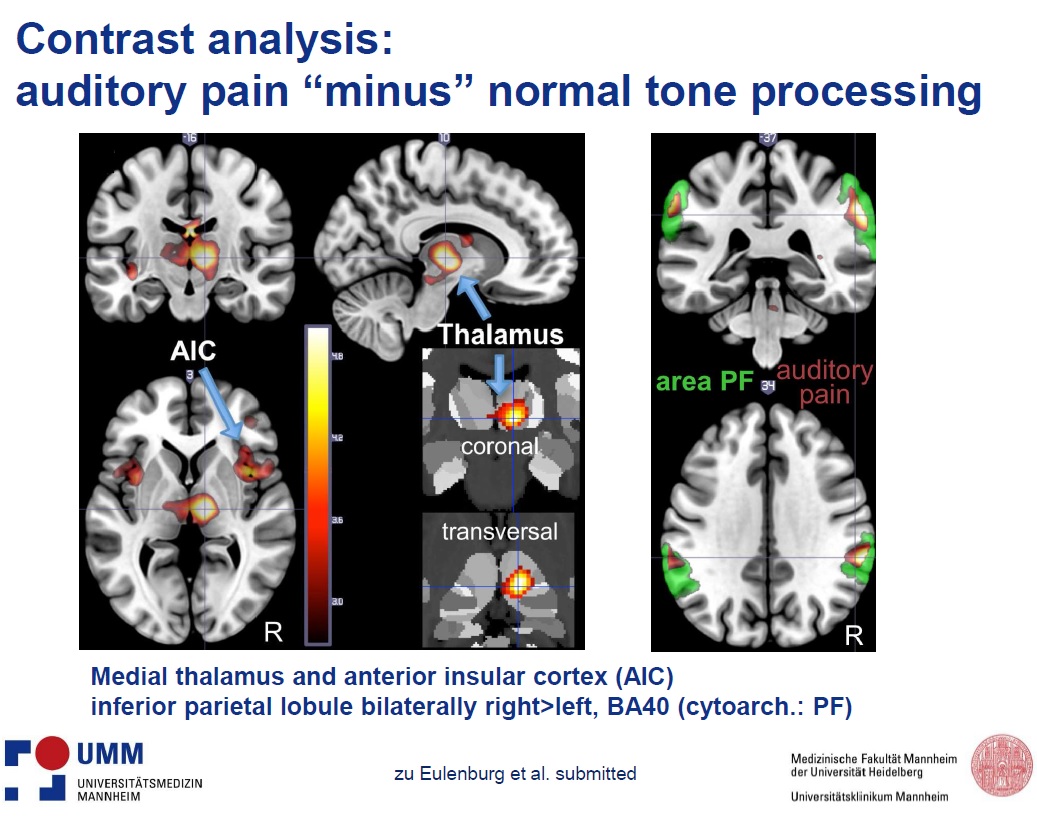
Conjunction analysis of painful auditory and pinprick stimuli showed activation of supramarginal and postcentral gyrus, inferior parietal lobule, rolandic operculum and left posterior insula, i.e. regions considered to be part of the nociceptive network in the brain. The strongest activation was seen in the Brodmann area 40, where nociceptive and visual convergence has been previously described.
Ulf concluded by stating that the “auditory pain” experience involves part of the network processing for somatic nociception and hence might be termed nociceptive. Since there was no pronounced activity in amygdala, hippocampus, or cingulate, it is less likely the process is solely a limbic-autonomic process, one without nociception.
The biology of neuropathic pain
Allan Basbaum, Professor of Anatomy, University of California, San Francisco

Allan Basbaum
While Allan had not previously investigated the nature of hyperacusis with pain, he synthesized the information from the other speakers and applied his expert knowledge of neuropathic pain to hyperacusis. He started by focusing on a few key descriptions of pain, such as “burning” and “stabbing.” These terms are commonly used by patients describing their neuropathic pain.
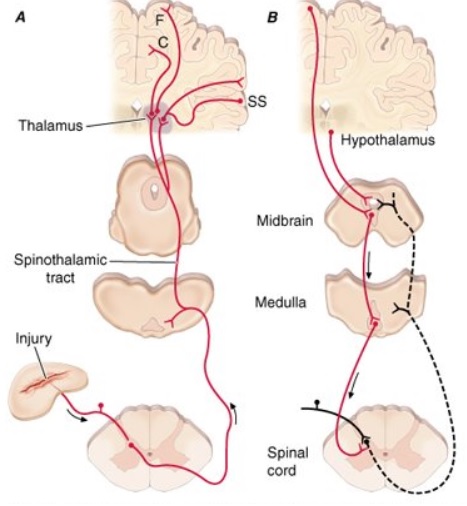 Allan first explained that the classic pain pathway description often given in textbooks (see this example) greatly oversimplifies the pain process. This pain process proceeds from an initial injury and a pain signal that is sent along either myelinated A-delta or unmyelinated C afferent fibers to the spinal cord and then to the thalamus in the brain (as shown in the right diagram). The A-delta and C nerve fibers respond maximally only to intense (painful) stimuli and produce the subjective experience of pain when they are electrically stimulated. Individual nociceptors can respond to several different types of noxious stimuli, such as heat, intense cold, intense mechanical stimuli such as pinch, and the application of irritating chemicals.
Allan first explained that the classic pain pathway description often given in textbooks (see this example) greatly oversimplifies the pain process. This pain process proceeds from an initial injury and a pain signal that is sent along either myelinated A-delta or unmyelinated C afferent fibers to the spinal cord and then to the thalamus in the brain (as shown in the right diagram). The A-delta and C nerve fibers respond maximally only to intense (painful) stimuli and produce the subjective experience of pain when they are electrically stimulated. Individual nociceptors can respond to several different types of noxious stimuli, such as heat, intense cold, intense mechanical stimuli such as pinch, and the application of irritating chemicals.
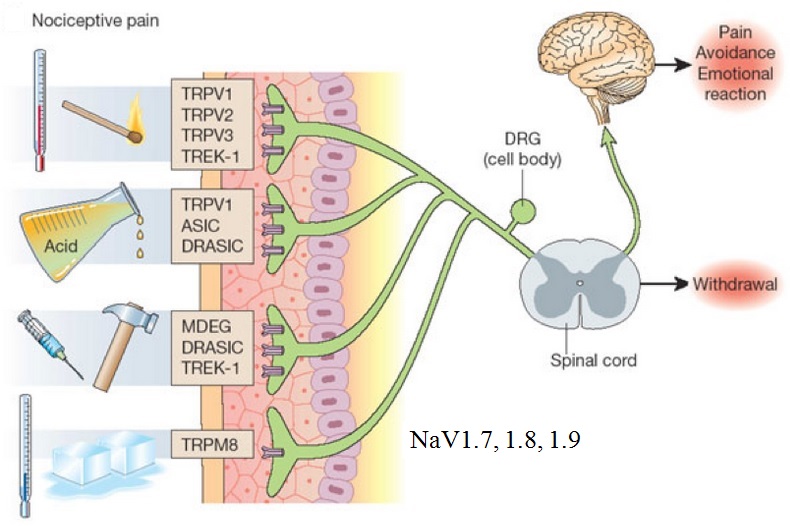
The basis of acute pain starts with one of many types of molecular transducers. For example, TRVPs that signal the presence of heat or TRPM8 that signals the presence of cold. (See image below from this pain site). NaV1.7 is a sodium ion channel that is expressed at high levels in the nociceptive (pain) neurons at dorsal root ganglion (DRG). In a genetic disorder where NaV1.7 is insufficient, a person may experience little or no pain (https://ghr.nlm.nih.gov/condition/congenital-insensitivity-to-pain). When there is a gain of NaV1.7, a person can experience excessive pain. Due to the significant role of NaV1.7 in the pain signaling process, a class of drugs targets this as a NaV1.7 inhibitor (see this article on NaV1.7 pain inhibitors).
Neuropathic pain typically is initiated from an injury to a peripheral site, although it can originate from the central nervous system. It is characterized by ongoing pain along with mechanical and thermal (often cold) hypersensitivity (allodynia and hyperalgesia). As local anesthetics are remarkably effective in producing at least temporary relief in many peripheral injury-induced neuropathic pain conditions, attention has focused on regulation of the afferent. Importantly, the primary afferent nociceptor expresses a host of molecules that are not found or are only minimally expressed elsewhere in the central nervous system.
Central sensitization of spinal cord pain-processing circuits also occurs after injury. NMDA-mediated central sensitization is manifest as increased ongoing activity of dorsal horn pain transmission circuits and their responsiveness to normally innocuous stimulation. The behavioral manifestation of these changes is ongoing pain, allodynia and hyperalgesia. Unfortunately, the widespread expression of the NMDA receptor has limited the development of selective antagonists to reduce pain because of adverse side effects. Allan’s view is that neuropathic pain is an epileptic-like condition resulting from reduced GABAergic inhibitory controls in the dorsal horn of the spinal cord. Consistent with this hypothesis, is the fact that anticonvulsants are among the most effective, or at least the most common, pharmacological approaches to treat neuropathic pain. Allan explained how some of these pharmacological approaches could be tested with severe hyperacusis patients. To date there is only anecdotal data on the effectiveness of any drug for relieving hyperacusis pain.
An alternative approach to reducing these pathophysiological consequences of injury is to re-establish the inhibitory tone lost in the setting of nerve injury. In recent studies, Allan has demonstrated that it is possible to restore inhibitory tone by transplanting GABAergic precursor neurons derived from the embryonic cortex into the spinal cord of nerve-injured mice. These precursor neurons develop in the spinal cord, differentiate into GABAergic interneurons and integrate into the host circuitry. Most importantly, peripheral nerve injury-induced mechanical hypersensitivity in a peripheral nerve injury model of neuropathic pain can be completely normalized within weeks of the transplantation.
The approach is also effective in the management of chemotherapy-induced mechanical and thermal (heat) hypersensitivity. With a view to translating these preclinical findings to patients, Allan has now initiated human studies using iPS cells modified to assume the properties of GABAergic neurons. Preliminary studies indicate that these cells have the capacity to integrate and influence host circuits. Taken together, these studies suggest that a therapy targeted at treating the “disease” of neuropathic pain, namely the pathophysiological alterations in CNS function that are characteristic of this condition, is a viable and novel approach to the management of neuropathic pain. Much work is needed to further investigate the cause of hyperacusis to know if this approach could also be used with hyperacusis.
A number of the key concepts discussed here can be studied more in depth in this paper of Allan’s: “Cellular and Molecular Mechanisms of Pain”. Also for a recorded lecture on the “The Science of Pain” see this video at UCTV.
Noise started bothering me after scanning geological logs at my former job. The hissing noise was just annoying at first, but for the last few years, any consistent noise (especially high pitched) bothers me. (Feels like a chopstick has been run thru my ears.) I had a brain tumor removed in 1983, and then part of my brain removed in 1992, to control seizures which started when I was nine months old. I am now 66, have been seizure free for at least ten years, but I have constant head and earaches from noise. I also have trouble sleeping.
I have profound hyperacusis and I suffer from CRPS(Complex Regional Pain Syndrome). Last Januaray,(2016), I received a nerve block to help my acute knee pain and I was told that at a later date my hyperacusis could be helped in the same way, but using another nerve. Unfortunately, the nerve bl0ck worsened my hyperacusis, lowering my threshold for sound sensitivity. Any thoughts. I am unable to live in the mainstream and my life has virtually become unmanageable. I am a retired Certified Music Therapist and musician who can barely tolerate the keys on my laptop.
If you use the “Contact Us” feature to send your contact information I’ll see if we know any specialists in your area.
My H is from very loud musics. This research shows only cure is to regenerate Type 1 and Type 2 cells and nerves. Hope they can or we have only pain for the rest of our lives. Thank you for research. I cant want for next meetings to learn more.
Hello im Really sorry you must be so busy but I was wondering if you could help give me any help at all. For 12 months now I have had a pain to short quick noises as low as putting my mobile onto the table or light switch or the every start of playing pink noise on my phone
I get pain face and head and my eye will close or I have sensations in the face
Or head.
Have had mra and MrI and it’s all normal
Could this be hyperacusis ?
Thank you so much
Best wishes and thank you for reading
Would anti seizure medications help with this condition? All
Developed after getting covid vaccine and made worse getting covid last winter. Any specialists you can recommend?