
Iver Juster, MD is a Family Medicine physician who trained and practiced for many years in the U.S. Subsequently he underwent additional training and work in U.S. managed care and medical informatics.[1] His work focuses on the design and evaluation of systems to support evidence-based clinical decision-making and tools that improve patient outcomes. In pursuing his desire to help a family member with hyperacusis he discovered that the best way to move our research agenda forward will be to bring all parties to the table in a meaningful way.

Bryan Pollard and Iver Juster, MD
Dr. Juster says: “On February 21, 2016, the Association for Research in Otolaryngology held a symposium on auditory nociception (the hearing system’s responses to potentially or actually harmful stimuli), focusing on pain in hyperacusis. The session convened researchers, clinicians, educators and—importantly—the voice of the patient—to coordinate our evolving knowledge of this disabling condition. The symposium was both intellectually and emotionally exciting, because we are finally recognizing the existence and importance of pain in hyperacusis, and are moving towards the understanding required for prevention and treatment.
“Characterization of the ‘Type II neuronal’ nociceptive pathway, the fact that when the cochlea is damaged or inflamed, it can be activated by moderate-level sound, and the possibility that the pain is neuropathic, holds considerable promise for individuals for whom ‘everyday’ sounds are painful. I take full responsibility for any factual errors and welcome comments. Opinions expressed here are my own.”
Pain is increasingly recognized as a part of hyperacusis for many individuals with this condition, but too many patients and clinicians would agree that it remains under-recognized, being lumped together with the broader characterization of hyperacusis as ‘annoyance’ or ‘intolerance’ to sounds.
Understanding the cause(s) and biology of pain in hyperacusis is important clinically because those with auditory pain may have a different type of hyperacusis, physiologically, compared to those who don’t. For example, such patients are often intolerant of sound generators, noting that they produce pain even at very low sound levels.
In this all-afternoon symposium six speakers presented various—yet importantly overlapping—facets of the biology of pain hyperacusis, including auditory nociception (the nervous system’s response to actually or potentially harmful stimuli). In brief:
- Rich Tyler and Bryan Pollard overviewed hyperacusis and its treatment from the patient’s and clinician’s perspective
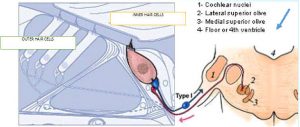
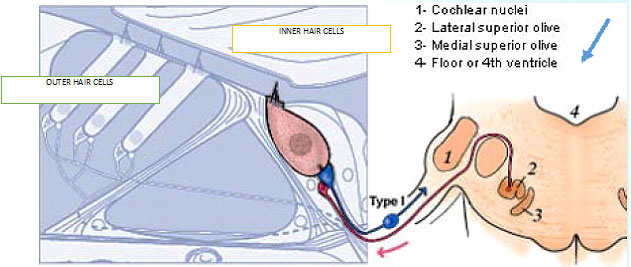
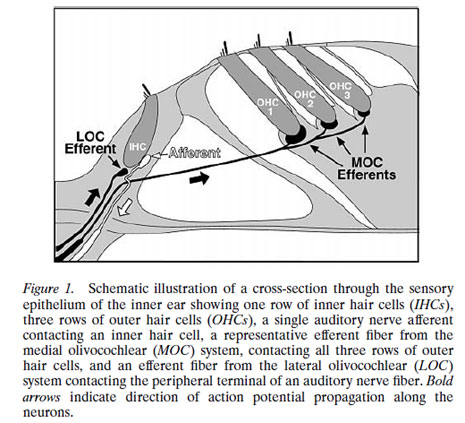
- Charles Liberman described the two types of “afferent” neurons that transmit signals from the cochlea: Type I neurons transmit information about sound (hearing); Type II neurons are nociceptive
- Jamie García-Añoveros led us further into the topic of auditory nociception and research that has revealed the central role and biology of Type II cochlear neurons
- Paul Fuchs showed us that damaged Type II neurons don’t require an extremely loud sound stimulus to transmit “this hurts!” information to the central nervous system
- Ulf Baumgaertner illuminated where in the brain sound and sound-related pain are processed, suggesting a way to visualize through fMRI what transpires in in the brain during painful hyperacusis
- Allan Basbaum suggested that at least in part, the pain of hyperacusis may be neuropathic; if true, this may allow us to tap into emerging major discoveries in treating this type of pain in patients with severe and disabling chronic pain
A deeper dive into each presenter’s talk follows. First the takeaways:
- Pain is an important part of hyperacusis for many individuals with the condition. Pain may be reactive to sounds near the loudness discomfort level (LDL), but often occurs with certain sounds at far quieter levels, such as whistles, blowers, tearing or rustling paper, or electrical equipment. Most tools used to evaluate the severity or life-impact of hyperacusis don’t get at this dimension, yet for many, pain with low intensity sound is disabling. The pain is often described as burning or piercing.
- The biology of hyperacusis pain has parallels to that of pain in most of the rest of the body. Stimulation of nociceptors by heat or intense pressure is served by different types of nerves than is light touch—the latter by thick myelinated nerve fibers; the former by smaller-diameter myelinated or by unmyelinated fibers. The two sensory systems travel through different pathways into and up the spinal cord to processing centers in the brain. A similar dichotomy serves touch versus nociception above the spinal cord, via the 5th, 7th, 9th and 10th cranial nerves.
 The biology of hyperacusis pain isn’t primarily due to stimulation of fibers in the 5th, 7th, 9th and 10th cranial nerves. These nerves serve parts of the eardrum (tympanic membrane), middle ear, and opening of the Eustachian tube in the throat, so some hypothesize that they mediate the experience of hyperacusis pain as well. Yet for the most part this doesn’t explain why ‘normal’-intensity causes pain.
The biology of hyperacusis pain isn’t primarily due to stimulation of fibers in the 5th, 7th, 9th and 10th cranial nerves. These nerves serve parts of the eardrum (tympanic membrane), middle ear, and opening of the Eustachian tube in the throat, so some hypothesize that they mediate the experience of hyperacusis pain as well. Yet for the most part this doesn’t explain why ‘normal’-intensity causes pain.- Anatomical studies offered a clue to the origin of hyperacusis pain via a ‘non-canonical’ pathway in the auditory nerve. Sensory information arising from stimulation of the cochlea is transmitted along the 8th cranial nerve. Auditory (sound) is translated by the cochlea’s inner hair cells (IHC) into impulses that activate thick myelinated “Type I” fibers that synapse (connect) on the IHC. This is the so-called ‘canonical pathway.’ The fibers’ cell bodies are located in the 8th cranial nerve’s spiral ganglion, and impulses carried by these nerves continue up to the brainstem, and thence to higher brain centers to be translated into what we perceive as sound. Type I fibers constitute 95% of the spiral ganglion.
But Type I fibers, synapsing on IHCs and carrying information about sound, are not nociceptive—that is, they don’t mediate the experience of pain. Type II fibers synapse on the OHCs and in people with normal ears they are activated—become nociceptive—only with extremely loud sounds (120 dB). The discovery of this “non-canonical pathway” means that under the right conditions the cochlea can be nociceptive, and that cochlear nociception doesn’t rely on the cranial nerves that serve sensation and pain in the rest of the head and neck. Instead, cochlear nociception is served by Type II fibers in the 8th cranial nerve. - If this was the entire story, we’d have arrived at a big “so what?” Hyperacusics who have pain complain of it at sound intensities far lower than 120 dB (the sound intensity that caused Type II fiber activation in experimental animals or pain with accompanying fMRI changes in experimental subjects)—many have reactive pain to certain sounds at 50 or 60 dB—often far lower than their LDLs.
- Based on research presented and discussed at the symposium, there may well be a big “so what.” We now enter the realm of speculation—but based on parallel observations in patients with neuropathic pain (a complex chronic pain state often accompanied by tissue injury, in which nerves send incorrect signals to pain centers. Like neuropathic pain, pain in hyperacusis is often described as burning, and triggered by what most people would regard as low-intensity stimuli. Neuropathic pain often occurs in an environment of chronic inflammation, and may sometimes be responsive to medications that increase the neurotransmitter, GABA. Viewed in this light, hyperacusis pain smells neuropathic.
- For many people with hyperacusis pain, this could be good news, because our understanding of the biology of neuropathic pain is exploding, and there look to be drugs on the horizon that could offer effective treatments. Even today, many hyperacusis patients find GABA-increasers like gabapentin (Neurontin®) and pregabalin (Lyrica®) helpful. Allan Basbaum described experimental treatments that may bear fruit in the next few years.
- In addition, to the extent that pain represents dysfunctional signaling from a chronically inflamed cochlea, perhaps emerging treatments for controlling out-of-proportion inflammation could be very helpful as well. Such treatments are currently used—or under investigation—with patients who have overachieving mast cells, the primary initiators and perpetrators of allergic and many intolerance-type responses.
_____________________________________________________
Diving deeper: Six perspectives on auditory nociception and pain hyperacusis
Note: Please see Bryan Pollard’s excellent summary at the link. What follows is an additional perspective on this excellent symposium. Appreciations to Bryan and Hyperacusis Research, and the presenters, for moving our understanding of this disabling condition forward.
Rich Tyler and Bryan Pollard overviewed hyperacusis, emphasizing perspectives of clinicians and patients. Hyperacusis: “An abnormal, excessive response to sound.” There are many known causes but most patients ascribe their hyperacusis to a sound injury. The genetics of hyperacusis is not characterized except in the rare William’s syndrome. (COMMENT: With the cost of whole-genome sequencing dropping exponentially, it’s time to explore the genetics of hyperacusis. Some patients may share predisposing metabolic abnormalities that can be addressed with diet, supplements, or medications)
Hyperacusis was categorized as: Loudness (sounds are perceived as louder than for a normal listener); annoyance (specific sounds); fear; and pain (where certain sounds evoke pain at modest intensities). Loudness H can be measured directly (LDLs); and there are standard questionnaires to get at the other types
COMMENT: What’s a useful categorization of hyperacusis? Terms should be chosen that have distinct, simple definitions based on criteria like etiology (cause), neuroanatomic pathways affected—and of importance for patients—consequences for treatment. In addition, there should be replicable, valid tests to diagnose or distinguish each type of hyperacusis—as is true in the rest of medicine.
People with hyperacusis often show sensitivity in other sensory systems such as being bothered by certain strong smells like cigarette smoke, bleach; or certain tastes). There’s a strong relationship with headaches; many also have balance problems or are bothered by bright lights.
Treatments: There are several; some help some people gain several dB of LDL but none have been found to come close to a cure:
- Medications: Anticonvulsants, benzodiazepines help some.[2] In evaluating effectiveness, it’s important to distinguish between reducing hyperacusis itself vs. patients’ reaction to having it. Other medications on the horizon may be useful if hyperacusis pain turns out to be neuropathic (see Allan Basbaum’s presentation below)
- Ear protection: Popular because it immediately reduces the noxious input, but by limiting exposure to sound could worsen hyperacusis. Richard Tyler generally recommends not using protection under normal sound circumstances and gradually weaning off protection for those using it. The evidence base for this is fairly weak however.[3] Yet, many patients do find that overprotection can worsen their sensitivity. Common sense, and ensuring one has sound protection at hand when really needed, seems reasonable.
- Hearing aids: Attenuating up to 30 db and offer control over how sounds are amplified[4]
- Sound therapy may use a continuous exposure to low level background noise.[5] A study by Craig Formby on patients with hearing loss showed LDLs can increase over several months with sound therapy.[6] Richard Tyler uses a different type of sound therapy for intolerance to specific sounds—successive approximation during relaxation. Counseling is often combined with sound therapies.[7] Sound therapy is not for everyone—some patients worsen. For more: http://hyperacusisfocus.org/research/soundtherapy/.
COMMENT: Not mentioned at ARO but of recent interest: Low-level laser therapy (“cold laser”) delivered to the cochlea through the external ear canal has some uncontrolled studies supporting several Db improvements in LDLs.[8][9] As a relatively inexpensive treatment with encouraging anecdotal support, it deserves a high-quality study. Oval and round window reinforcement, an outpatient surgery used in patients with superior canal dehiscence syndrome, aims to stiffen the cochlear apparatus, thus transmitting less energy to the sound-receiving inner hair cells. Early results for patients with hyperacusis look promising, and clinical trials have begun.[10] Until we better understand the biology of hyperacusis, most patients will require a multi-dimensional treatment program customized to their situation and preferences.
_____________________________________________________
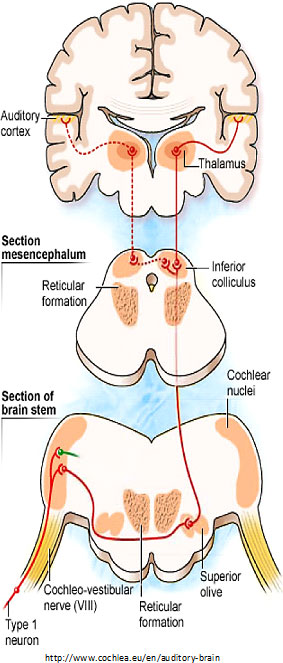 In “The cochlear nerve: Type I vs. Type II neurons and the phenomenon of hyperacusis,” Charles Liberman
In “The cochlear nerve: Type I vs. Type II neurons and the phenomenon of hyperacusis,” Charles Liberman
described the two types of “afferent” (inbound) neurons that transmit signals from the cochlea: Myelinated Type I neurons transmit information about sound (hearing) from the cochlea’s inner hair cells (IHC); Unmyelinated Type II neurons are nociceptive (respond to potentially harmful stimuli like heat and heavy pressure) and carry information from the cochlea’s outer hair cells (OHC).
Both Types project to the cochlear nucleus (CN) and bifurcate. The diagram shows the journey of Type I neurons—the classical afferent information pathway for the auditory branch of the 8th cranial nerve.
Type II fibers in the central nervous system are so thin that they weren’t seen until the 1980s using electron microscopes.
Type I IHC synapses are big and fast communicators, each with hundreds of synaptic vesicles (containing chemicals—neurotransmitters—that transmit information from one neuron to the next). Each connects with a single IHC. Type II OHC synapses are slow, harder-to-activate communicators with fewer synaptic vesicles that connect with many OHCs.
Furthermore, Type II neurons may have reciprocal synapses capable of both afferent and efferent communication. This may allow OHCs to talk with each other. Read more about Type II neurons at the end of this article.
_____________________________________________________
In “Auditory nociception, the detection of sounds harmful to the inner ear, is mediated by a non-canonical form of communication from cochlea to brain,” Jamie Garcia-Aňoveros
invited us further into the topic of auditory nociception and research that has revealed the central role and biology of Type IIs.
He opened with the observation that intense, persistent noise will damage and ultimately destroy cochlear hair cells. But—unlike most of the rest of the body where nociceptors of the dorsal root and trigeminal (5th cranial nerve) ganglia detect tissue damage (or the physical stimulus causing it)—somatosensory neurons don’t innervate the organ of Corti. He posed the question: Does the cochlea have a nociceptor like mechanism like the rest of the body does?
We know that with sound stimulation, IHCs release the neurotransmitter glutamate to activate Type I neurons’ AMPA receptors, which carry signals to the lower brain’s cochlear nucleus (you can see this route diagrammed above with Charles Liberman’s talk). But what’s the function of Type II neurons, which are unmyelinated, contact OHCs, and make up 5% of the spiral ganglion?
What follows is somewhat technical, but sheds light on the painstaking, disciplined brilliance of research into hyperacusis and hearing. Lab mice can be bred to “knock out” a single gene among all the genes carried in their DNA. What’s actually knocked-out is the normally-occurring (“wild”) version of the gene; it’s not as if the part of the chromosome that gene lives on is replaced by a hole! The wild, functional DNA is replaced with a non-functioning gene.[11] Since genes are responsible for the production of proteins, and proteins have various biological functions, rendering a specific gene non-functional will cause its protein product to not be produced (or to be produced but be non-functional). A mouse bred to have a non-functional gene is called a knock-out (KO) mouse.
Jaime and his team investigated the question in Vglut3-/- KO mice[12], which are bred to have the wild Vglut3 gene knocked out. These KO mice don’t produce the Vglut3 protein, which is involved in the IHC AMPA receptor for the neurotransmitter glutamate—the starting point for the canonical auditory pathway. No AMPA means no activation of Type I neurons—in other words, the canonical pathway of Vglut3-/- KO mice is silenced; the mice are deaf.
Despite this silencing, they documented neuronal activation in the brainstem in response to noxious noise—consistent with the activation of Type II afferents which does not depend on glutamate:
- Noxious (but not sub-noxious) noise[13] damages hair cells, and they found responses to noxious—but not sub-noxious—noise in the cochlear nucleus (CN).
- They did not find responses to noxious noise in the trigeminal (5th cranial nerve) or vestibular (part of the 8th cranial nerve serving balance) nuclei.
- Noxious noise activated CN neurons in a pattern consistent with the innervation of Type II afferents. In addition, Type II neurons express proteins we’d expect to find in nociceptors, such as peripherin.[14]
His conclusion: The response to noxious noise starts in the cochlea—not in middle ear or vestibule, and information about it is transmitted from OHCs via Type II auditory neurons. Jaime used the term noxacusis to refer to activation of the Type II afferent-mediated non-canonical pathway.
_____________________________________________________
In “What is the ‘adequate stimulus’ for type 2 cochlear afferents?” Paul Fuchs
showed us that damaged Type II neurons don’t require an extremely loud sound stimulus to transmit “this hurts” information to the central nervous system.
Paul overviewed the nature of Type II afferents: they are thin unmyelinated neurons that extend hundreds of microns along the cochlear duct to contact many OHCs—in contrast to Type Is which contact only a single IHC. Type IIs and the OHCs are not designed to deliver auditory information. But they’re strongly activated when OHC are damaged. This response depends on purinergic receptors (which recognize ‘purine’ neurotransmitters like adenosine triphosphate—ATP). When OHCs are damaged, their supporting cells release ATP, and the purinergic receptors bind the ATP. This implies that Type IIs may be the cochlea’s nociceptors whose purpose is to prompt avoidance of further damage from loud sounds.
He described an experiment[15] that supported this; the researchers hypothesized this as a potential mechanism for pain in hyperacusis.
COMMENT: Other investigations have shown that Type II afferents convey signals leading to the perception of pain only when stimulated by extremely loud sound. But that research was done on animals and humans with healthy cochleae. Paul’s findings suggest that when OHCs are damaged, Type II fibers become easier to activate, leading to a reduced threshold for nociception. This may tie in with our emerging understanding of neuropathic pain and the maintenance of chronic pain generally: When nociceptors are damaged or situated in a chronically inflamed local environment, they participate in positive-feedback loops that maintain nociceptive sensitivity (as described below).
_____________________________________________________
In “Auditory vs. aural nociception: Evidence for partial overlap of cortical activity in humans,” Ulf Baumgaertner
illuminated where in the brain sound and sound-related pain are processed, suggesting a way to visualize through fMRI what transpires in hyperacusis.
The somatosensory system has 2 pathways to transmit information about touch, temperature, position and pain. The pathways are served by different afferent nerve fibers.[16] These pathways are discussed in more detail (with a diagram) below, in Charles Liberman’s talk.
- Low activation-threshold for touch and proprioception—position in space: large-caliber fibers, entering the dorsal column of the spinal cord and projecting through the medial lemniscus
- Higher activation-threshold for temperature and nociception: small caliber (A-delta and C) fibers carried in the anterior spinothalamic tract
As these pathways ascend the central nervous system, they increasingly interact.
Ulf asked how an intense auditory stimulus might activate the nociceptive system. In theory activation could arise from excitation of trigeminal (5th cranial nerve) nociceptive fibers of the middle ear; or might be an unpleasant feeling unrelated to nociception via a standard sensory channel from inner ear cells converging on limbic areas (amygdala, cingulate) or could be a separate high-threshold sensory channel.
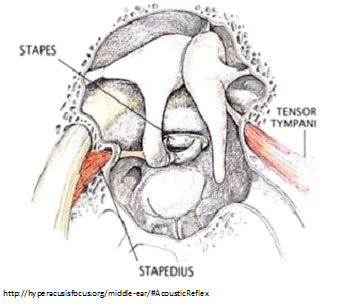 Trigeminal nerve isn’t likely, because its afferents are not excited by sound frequency.
Trigeminal nerve isn’t likely, because its afferents are not excited by sound frequency.
(COMMENT: There are two middle-ear muscles that control the impact of intense sound: the tensor tympani and the stapedius. Could spasms of these muscles be a source of pain in hyperacusis? Not likely: Tensor tympani spasm—myoclonus—results in pulsing tinnitus; and since stapedius contracts during speech, we’d expect that if it was a source of pain, we’d notice it when talking. Nonetheless, some hyperacusics say they can feel tender tightness in muscles underneath their outer ear—behind the jaw—or in the chewing muscles, when exposed to certain sounds. Perhaps this specific type of pain arises from some of the many muscles controlling the jaw and temporomandibular joint).
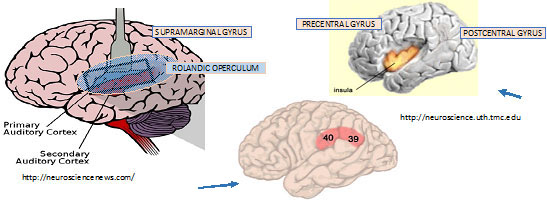 To return to our story, Ulf cited an fMRI study on healthy volunteers using auditory and somatic stimulation. Two levels of auditory stimulation were applied: 3200 Hz at 130 dB (perceived as painful by all subjects); and 3200 Hz at 75 dB. Subjects perceived the intense auditory stimulus and the earlobe stimulus as equally painful.
To return to our story, Ulf cited an fMRI study on healthy volunteers using auditory and somatic stimulation. Two levels of auditory stimulation were applied: 3200 Hz at 130 dB (perceived as painful by all subjects); and 3200 Hz at 75 dB. Subjects perceived the intense auditory stimulus and the earlobe stimulus as equally painful.
On fMRI:
- Both painful and non-painful auditory stimuli showed activation in the same areas in the brain—the primary and secondary auditory cortices.
- If stimulus was perceived as painful, they saw more activity in medial thalamus, anterior mid-insular cortex bilaterally (both sides) and inferior parietal lobule bilaterally—Brodmann area 40.
- Similar areas of the brain were de-activated with both auditory and sensory painful stimuli.
Overall, the 2 types of painful stimuli activated some brain areas in common, but with some areas of activation unique to each. The experiment strongly suggested that high-intensity auditory stimuli, which we are characterizing as being conveyed centrally starting with Type II cochlear neurons, exhibits the brain pattern characteristic of nociception. The involvement of Brodmann Area 40 is noteworthy: This large area of the brain serves sensory integration—for example somatosensory with visual, and auditory.
During Q&A, an audience member mentioned that there are projections from the trigeminal ganglion to the cochlea and into blood vessels – perhaps autonomic (see below); participation of the trigeminal nerve hasn’t been ruled out as part of the hyperacusis pain experience.
COMMENT: The more we know, the more we know that we need to know more.
_____________________________________________________
In “The biology of neuropathic pain,” Allan Basbaum
suggested that at least in part, the pain of hyperacusis may be neuropathic; if true, this may allow us to tap into emerging major discoveries in treating this type of pain in patients with severe and disabling chronic pain. I’d like to dive fairly deep into Allan’s presentation because it offers a novel (and potentially important) way of looking at hyperacusis pain, and as a way of tying together major themes of this memorable afternoon.
Allan applied his expert knowledge of neuropathic pain to pain in hyperacusis. Based on hyperacusics’ descriptions of their pain—burning, stabbing and out of proportion to the intensity of the stimulus—he invited us to consider that hyperacusis pain may be neuropathic.[17] This model is intriguing given other investigators’ findings that for a Type II fiber to transmit the information “this is a noxious stimulus” requires sound intensities far greater than those that typically cause reactive pain in people with hyperacusis.
The hypothesis that pain hyperacusis is at least in part neuropathic could connect this perplexing condition to recent exciting discoveries with potential of substantial relief for people with neuropathic pain, such as complex regional pain syndrome.[18]
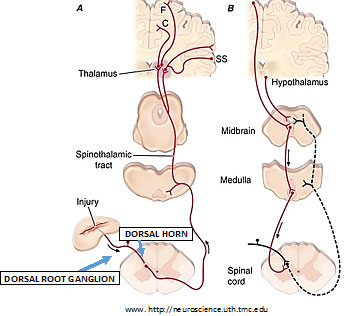 He set the stage with a review of the classical neuroanatomy of pain. The primary sensory afferent peripheral nerves carry messages about light touch, motion, temperature and pain to the spinal cord. There are three types of primary sensory afferents:
He set the stage with a review of the classical neuroanatomy of pain. The primary sensory afferent peripheral nerves carry messages about light touch, motion, temperature and pain to the spinal cord. There are three types of primary sensory afferents:
- A-beta: Large-diameter myelinated; carry messages about light touch and motion
- A-delta: Small-diameter myelinated; they respond to noxious stimuli such as intense mechanical stimuli, temperature, and irritating chemicals including ATP, serotonin, bradykinin and histamine
- C: Small-diameter unmyelinated; they respond to the same stimuli as A-delta.
A-delta and C fibers respond maximally only to intense stimuli; they’re called nociceptors (pain receptors). In nerves that enter the spine, sensory messages travel along the afferents through cell bodies located in the dorsal root ganglion to synapse in the spinal cord, and terminate in the dorsal horn.
There they release neurotransmitters—fast-acting glutamate and slower/longer-acting substance P; and calcitonin gene-related peptide (CGRP) that excite neurons in the dorsal horn, most of which cross the midline to project to the contralateral (other side) thalamus, as the spinothalamic pathway.
The spinothalamic tract neurons synapse again in the thalamus and ascend to many areas in the cerebral cortex—serving different facets of the experience of pain, such as location, intensity, quality (somatosensory cortex), and emotion (cingulate gyrus, insular cortex and other areas in the brain’s frontal lobes).
 In addition to this afferent pathway there is a pain-modulation pathway, where inputs coming down from the hypothalamus and frontal cortex activate cells in the midbrain that control spinal pain-transmission cells via synapses with cells in the medulla. This means that the afferent sensory pathway transmitting information up the spinal cord and brain interacts with efferent information (or feedback) coming down from the brain.
In addition to this afferent pathway there is a pain-modulation pathway, where inputs coming down from the hypothalamus and frontal cortex activate cells in the midbrain that control spinal pain-transmission cells via synapses with cells in the medulla. This means that the afferent sensory pathway transmitting information up the spinal cord and brain interacts with efferent information (or feedback) coming down from the brain.
This efferent pathway is responsible for a variety of responses to noxious stimuli and is thought to contribute to the placebo response and mental pain control as well as the effect of exogenous (e.g. medications or plant substances) or endogenous (made in the body) opioids on pain.
How sensory information may become neuropathic: When nociceptors become damaged or exposed to prolonged stimulation or inflammation, they become sensitized—activated with only mild or even no stimulation. Where what were once innocuous stimuli now produce pain. Everyone has experienced sensitization as the tenderness following a sunburn or with a patch of inflamed skin. Sensitization is not just limited to skin—it can occur in joints or along the gastrointestinal or urinary tracts.
In fact, it’s been shown that afferent nociceptors can themselves contribute to inflammation when activated by releasing polypeptides (molecules consisting of a small number of amino acids). For example, when substance P (an 11-amino acid polypeptide) is released from primary afferent nociceptors, it results in vasodilation, mast-cell degranulation, attracts leukocytes, and promotes production and release of inflammatory mediators.[19]
This is thought due in part to an increase in the number of sodium channels. Sensitization occurs when the central (spinal cord and above) nervous system can become involved—as does the sympathetic nervous system, as the damaged nociceptors become highly sensitive to epinephrine and norepinephrine—thus become activated by the efferent sympathetic nerves that travel along with the peripheral afferents.
Important phenomenon: If inflammation is present, sympathetic activity can activate undamaged nociceptors.
This raises the question of whether inflammation—with its release of inflammatory chemicals—may be important to initiating and maintaining pain in hyperacusis and whether hyperacusis pain is neuropathic. Experiments showing that Type II cochlear neurons can be activated by very intense sound were conducted in animals and humans free of hyperacusis; but perhaps in hyperacusics, the initiating processes and chronic inflammation result in sensitization with the transmission of pain signals with relatively low-intensity sounds—especially sounds of certain qualities that are difficult for the damaged or inflamed cochlea to process—such as tearing paper, leaf-blowers or electrical equipment.
A research avenue with exciting potential is targeting the ‘voltage-gated sodium channels’ of nociceptors. These are labeled Nav (Na is the chemical symbol for sodium), and there are many types throughout the body; nociceptors express types 1.7, 1.8 and 1.9. Nav1.7 is the most interesting—a gain of Nav1.7 function produces neuropathic pain and erythromelalgia, a neuropathic inflammatory response involving massive vasodilation. Loss of function of Nav1.7 results in a loss of sensitivity to pain. Research is needed to discover which voltage-gated channels are expressed in the Type II neurons.
Possible treatment implications:
- One way to get more input from same stimulus is to get more release of the same neurotransmitter – this is due to calcium flux. This is why gabapentinoids are effective; they modulate calcium channel binding. Gabapentinoids like gabapentin (Neurontin) and pregabalin (Lyrica) are first line choices in neuropathic pain, but only 30% effective in 30% of patients.
- NaV1.7 inhibitors could produce a highly selective block. This is a hot target for pharmaceutical companies now because the target is restricted to C-fiber presynaptic terminals. Allan commented that tricyclic antidepressants (which have been around for many decades) are good Na channel blockers and are typically used as second-line treatment
- Postsynaptic side: Increased response to a transmitter strengthens synaptic excitability and efficiency; this occurs in receptors for neurotransmitters NMDA and AMPA. There are no acceptable NMDA receptor blockers; ketamine is sometimes used though often limited by side-effects.
- A major contributor to neuropathic pain is development of quasi-epileptic foci due to loss of GABAergic, glutaminergic and glycinergic inhibitory interneurons (GABA, glutamate and glycine are neurotransmitters). So loss of GABAergic inhibitory interneurons is important – there is less inhibition and more excitability. In this situation with epilepsy we would aim to build up the lost inhibitory tone with gabapentinoids and other anticonvulsants.
- Yet another factor appears to be activation of microglia induced by nerve injury. Microglia are scavenger cells located throughout the brain and spinal cord. They form the first line of immune defense for the central nervous system. Cutting a peripheral nerve induces massive proliferation of microglia at the injury site. An essential part of the microglial defense is to disgorge molecules that call in other immune cells and initiate inflammation. With inflammation comes neuronal irritability—a hallmark of neuropathic pain.
Treatment of neuropathic pain includes topical anesthetics, nerve blocks, TENS, gabapentinoids, serotonin-norepinephrine reuptake inhibitors (SNRI, often used as antidepressants). Opioids (like Vicodin®) are not very effective in neuropathic pain.
In response to a question from the audience about cannabinoids (chemicals found in marijuana that stimulate two known types of receptors, CB1 and CB2 and that mimic the body’s own endocannabinoid system), Allan said the topic is controversial and needs more (or higher quality) research.[20]
Next Allan described a fascinating experiment in mice, illustrating a potential approach to targeting one of the contributors to neuropathic pain: the loss of inhibitory signaling. In this approach the missing GABAergic neurons are replaced by transplanting embryonic GABAergic precursors into the spinal cord. Then, GABAergic nerve transplantation was tried in humans and they “took.” Back to mice, the transplanted neurons spread around the cord and integrated into its central nervous system. Further, the transplants were able to reduce induced pain of complex regional pain syndrome—suggesting a repair of the central nervous system.
For the very curious: More about Type II neurons
 AMPA is a cell membrane receptor for the neurotransmitter glutamate, which mediates fast synaptic transmission. Type II neurons’ synapses with OHCs don’t appear to express AMPA receptors (Liberman, 2011). This means they do not respond to the neurotransmitter glutamate (the neurotransmitter recognized by Type I neurons in their synapses with IHCs). Unlike Type Is—which innervate a single IHC apiece, Type IIs can innervate many OHCs, and they respond only when OHCs are damaged. This situation can occur with extremely loud sounds, as in the experiments described by Paul Fuchs and Ulf Baumgaertner
AMPA is a cell membrane receptor for the neurotransmitter glutamate, which mediates fast synaptic transmission. Type II neurons’ synapses with OHCs don’t appear to express AMPA receptors (Liberman, 2011). This means they do not respond to the neurotransmitter glutamate (the neurotransmitter recognized by Type I neurons in their synapses with IHCs). Unlike Type Is—which innervate a single IHC apiece, Type IIs can innervate many OHCs, and they respond only when OHCs are damaged. This situation can occur with extremely loud sounds, as in the experiments described by Paul Fuchs and Ulf Baumgaertner
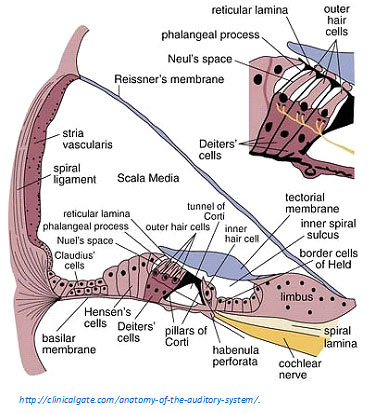
Type IIs innervate Hensen’s Cells in the apical cochlea. Hensen’s cells form a buttress for other structures in the cochlea. There are many Type II bundles traveling from the third row of OHCs to the Hensen’s cells in the apex; they are talking to the Hensen’s cells, not receiving info from Hensen’s cells.
The relevance of this to hyperacusis pain is that Hensen’s cells collapse and recover when overstimulated in vitro (in the lab; in vivo refers to in the body). This dynamic process could be triggered by the nociceptors, telling Hensen’s cells to collapse the Hensen buttress to reduce mechanical stimulation from loud sounds.
(COMMENT): what are the implications for the sound intensities at which hyperacusis pain is typically triggered?)
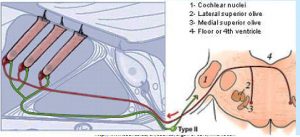
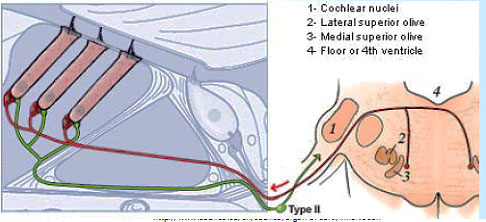
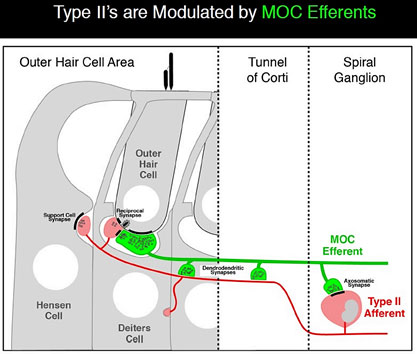
 Type IIs are modulated by medial olivocochlear (MOC) efferents, which synapse with Type IIs in the OHC, and on Hensen and Deiter’s cells. Liberman and Maison published on this “MOC reflex” in 2000.[21] The illustration is taken from that paper.
Type IIs are modulated by medial olivocochlear (MOC) efferents, which synapse with Type IIs in the OHC, and on Hensen and Deiter’s cells. Liberman and Maison published on this “MOC reflex” in 2000.[21] The illustration is taken from that paper.
Several studies about the MOC reflex have been published since, exploring its neurobiology and genomics; but not all authors are satisfied that the reflex is important in the experience of auditory pain. A study published in 2015 looking at peripherin knockout mouse, found that Type II drive the MOC reflex. Peripherin is a good marker for Type II cell bodies in the normal animal,[22] but the knockout mouse showed no uptake by Type II cell bodies. However, the MOC reflex is supposed to be a sound mediated loop. If Type II cells are driving it, they’d have to do it at 20 db and using nearly as short an action potential latency as if the reflex was driven by Type I. This is against much of what we knew about physiology. However, in a to-be-published study, Maison and Liberman found that Type II are NOT absent in peripherin knockout mice, thus concluding that dysfunction in the MOC reflex is due to efferents.
Footnotes
[1] The Health Information and Management Systems Society defines medical informatics as “the interdisciplinary study of the design, development, adoption and application of information technology-based innovations in healthcare services delivery, management and planning.” Much is missing from this technology-oriented perspective, such as the study of how clinicians, patients/families, doctors, and society make clinical decisions; and on the importance of valid methods of evaluating the effect of diagnostic tests and treatments.
[2] Such as risperidone (Risperdal—used as an antipsychotic); the anticonvulsants levetiracetam (Keppra®), retigabine (Ezogabine®), gabapentin (Neurontin®) and pregabalin®), and benzodiazepines (e.g. Valium® and Xanax®)
[3] Most studies have been on normal hearing patients without hyperacusis who experienced several dB LDL loss after wearing ear protection for several days (Formby).
[4] High level sounds are peak-clipped or compressed; adjusting input/output (gain) so low level sounds are amplified but not the high level…followed by gradual transition to “normal”
[5] Hazell JWP, Sheldrake JB. Hyperacusis and tinnitus. In: Aran J-M, Dauman R, ed. Tinnitus 91: Proceedings of the Fourth International Tinnitus Seminar. Amsterdam/New-York: Kugler Publications, 245–248, 1992.
[6] Formby C, Hawley M, Sherlock L, Gold S, Payne J, Brooks R, Parton J, Juneau R, Deporte E, Siegle G. A Sound Therapy-Based Intervention to Expand the Auditory Dynamic Range for Loudness among Persons with Sensorineural Hearing Losses: A Randomized Placebo-Controlled Clinical Trial. Seminars in Hearing 2015:36(2):77-109.
[7] Described for tinnitus at https://hearinghealthfoundation.org/index.php?pg=tinnitus_counseling_sound_therapy.
[8] Prosper J, Graffelman J. The analysis of audiometric measurements before and after low-level laser therapy of Spanish patients with hyperacusis. (White paper report, 2013)
[9] Zazzio M. Pain threshold improvement for chronic hyperacusis patients in a prospective clinical study. Photomed Laser Surg 2010;28(3):371-377.
[10] Silverstein H, Wu YE, Hagan S. Round and oval window reinforcement for the treatment of hyperacusis. Am J Otolaryngology 2015;36(2):158-162.
[11] For more on KO mice, see https://www.genome.gov/12514551/knockout-mice-fact-sheet/.
[12] Vglut3 is the 3rd type of vesicular glutamate transporter to be discovered. Mice with Vglut3 knocked out are deaf.
[13] The researchers compared KO and wild mice in three experimental conditions: No noise, 80 dB noise (considered non-noxious from a normal cochlea’s point of view) and 120 dB noise (noxious – meaning that it can cause observable physical damage). With noxious noise, OHCs were destroyed and there was also some injury to the ICH. In the Vglut3-/- mice—without a functioning Type I pathway—non-noxious noise didn’t activate the cochlear nucleus, but noxious noise did, proving that noxious-noise information must be transmitted by a non-canonical pathway—that is, via Type II neurons.
[14] Flores EN, Duggan A, Liberman MC, Garcia-Aňoveros J. A non-canonical pathway from cochlea to brain signals tissue-damaging noise. Current Biology 2015;25;606-612.
[15] Liu C, Glowatzki E, Fuchs PA. Unmeylinated type II afferent neurons report cochlear damage. Proceedings of the National Academy of Sciences 2015;112(47)14723-14727.
[16] Treede RD. Neurophysiological studies of pain pathways in peripheral and central pain disorders. J Neurol 2003;250(10):1152-1161.
[17] The American Chronic Pain Association defines neuropathic pain as a complex, chronic pain state that usually is accompanied by tissue injury…nerve fibers themselves might be damaged, dysfunctional or injured. These damaged nerve fibers send incorrect signals to other pain centers. https://www.theacpa.org/condition/Neuropathic-Pain.
[18] The NIH National Institute of Neurological Disorders and Stroke: “Complex regional pain syndrome is a chronic pain condition most often affecting one of the limbs, usually after an injury or trauma to that limb. CRPS is believed to be caused by damage to, or malfunction of, the peripheral and central nervous systems…. CRPS is characterized by prolonged or excessive pain and mild or dramatic changes in skin color, temperature, and/or swelling in the affected area. There are two similar forms, called CRPS-I and CRPS-II, with the same symptoms and treatments. CRPS-II (previously called causalgia) is the term used for patients with confirmed nerve injuries. Individuals without confirmed nerve injury are classified as having CRPS-I (previously called reflex sympathetic dystrophy syndrome). Some research has identified evidence of nerve injury in CRPS-I, so the validity of the two different forms is being investigated.
[19] Events leading to activation, sensitization and spread of sensitization of primary afferent nociceptor terminals: PRIMARY ACTIVATION: (1) Direct activation by intense pressure; (2) consequent cell damage; (3) reduced pH leads to (4) potassium release and synthesis of prostaglandins and bradykinin. (5) Prostaglandins increase sensitivity of the nerve terminal to bradykinin and other pain-producing substances. SECONDARY ACTIVATION: (6) Impulses generated in the activated nerve terminal propagate to spinal cord and other terminal branches (7) where they induce release of peptides, including substance P, which (8) causes vasodilatation and neurogenic edema and further accumulation of bradykinin, (9) release of histamine from mast cells, and (10) serotonin (5HT) from platelets.
[20] For a recent review of the topic, see Fine PG, Rosenfeld MJ. Cannabinoids for neuropathic pain. Curr Pain Headache Rep 2014;18:451-460.
[21] Maison S, Liberman MC. Predicting vulnerability to acoustic injury with a noninvasive assay of olivocochlear reflex strength. J Neuroscience 2000;20(12):4701-4707.
[22] Froud KE, Wong ACY, Cederholm JME, et al. Type II spiral ganglion afferent neurons drive olivocochlear reflex suppression of the cochlear amplifier. Nature Communications 2015; DOI: 10.1038/ncomms8115.
Speaking as an admin for Hyperacusis Sufferers FB group, I think this is the single most important piece of literature that I have ever seen about Hyperacusis.
It was so thorough that it gave a complete “state of the union”, but it can also be used as a tool for patients to educate their doctors including primary care, and could be highlighted to show just a few important facts to friends and family members.
I am absolutely in tears. I feel like I’ve been waiting 11 years to read this article. I thank anyone involved in the research going on, and I give grateful thanks to Dr. Iver Juster for this incredible technical report that is so well-written, well organized, and thorough, that I was able to understand without a medical background. Thank you. Thank you.
Yes, to the “I’m in tears” comment.
This is the most hopeful report I’ve ever seen on hyperacusis.
I’ve had three years of agony.
Maybe there’s hope for a cure—in my lifetime.
Thanks for making this detailed report available.
This is a great article. My worst experience with the medical fraternity here is to find that l am not even believed when l try to describe my symptoms, and then find myself being told to consult a psychiatrist.
I am curious about the descriptions of hyperacusis pain as burning and/or stabbing as l don’t experience that. For me the pain/discomfort (depending on the intensity) is not precisely in my ears but more generally in my head. It’s so difficult to describe, but it is more like a tightening sensation throughout my head like claws in my brain. It is rather like what normal people experience when exposed to loud microphone feedback sound. Also, l feel very bad when exposed to sudden changes of volume. A sudden increase in volume for me feels as if l have been punched in the head by a sound wave through my ears. If l am exposed to music with a strong rhythmic aspect, staccato or short bursts of sound, it’s like continuous punching and l cannot tolerate it for long. Frequency also is critical, higher and lower frequencies being worse than midrange frequencies. I suffer more with motorbike engines, compression brakes and boom boxes than general continuous white noise traffic sounds. Screeching brakes, the many “ding dongs” and beeps etc from public PA systems and microwave ovens are also troublesome, but the sensation they cause is neither burning nor stabbing, yet it is a distinct physical discomfort. I don’t think it has anything to do with fear or annoyance and is definitely directly related to the volume as well as frequency spectrum. Overall, l feel as if your description does not quite fit my case. Are there others like me or am l alone?
EH—You’re certainly not alone. Most of the descriptions of the pain you experience resonate (bad verb) all too well with me. Feeling “claws in my brain,” or getting “punched in the head,” or “continuous punching”: yes to all three.
The triggers you describe are also all too familiar: “ding dongs, “bings,” “microwave ovens,” and especially amplified sound. I’m now afraid of microphones, loud speakers, telephones, car horns, police sirens, and more. Electronic music, except at rather low levels, is intolerable. A gun shot, even at some distance—horrible. A single acoustic shock leaves me trembling. A severe acoustic shock can render me helpless for hours or
days. Then even quiet conversation becomes unendurable.
I used to describe my pain as “hot knives through the ear into my brain.”
Then I added, “like a baseball bat against the side of my head.”
Now I add, “like being electrocuted.”
Some days I almost forget I have hyperacusis.
Other days, its almost the only thing I can think about.
Thanks for writing.
I know someone who is been recently diagnosed with Hyperacusis. She is still attending education and is struggling to say the least to keep up with her studies. As an educator I would love to know if there is something I could/should do to support this person. Any advise is tremendously appreciated.
Thank you.
The most important step a friend can take is to acknowledge the the painful realities of the condition. We still have a lot of work to do to educate the clinical community about how devastating hyperacusis can be so it is important the anyone with hyperacusis has a good support system. Getting educated yourself by studying the research we feature on the site can be helpful in providing support.
Further to my previous comment, l forgot to add that in addition to the above, l also experience popping and crackling of my eardrums and also occasional rapid stapedial ticking on exposure to sudden sounds. Sometimes the sensation of aural fullness is very strong and l have to use the Valsalva manoeuvre to relieve it but it was not always successful and sometimes had unpleasant effects of making me feel temporarily deaf even though my hearing is normal. These things are very annoying but not painful. The popping and crackling can occur in response to my own voice as well as external sounds, making conversations by phone difficult. My overall sensitivity has decreased since onset 8 years ago, but it still fluctuates from day to day and throughout the day. I just wonder if this is typical. I thought they were fairly common but l note that these aspects of hyperacusis have not been mentioned in the summary of the ARO proceedings.
We do try to track all types of symptoms. This is one of many purposes of the Sanford Hyperacusis Research Survey. Aural fullness and ear drum ‘flutter’ or ‘popping’ are very common. We do hear from some who have effects or pains in various parts of their head but as indicated this is often difficult to fully describe.
Excellent article. Great to see some serious research going on.